UPDATE: Frissítésként Sarah Walker (a plakát társszerzője) válaszolt a kérdésemre a GATK fórumon. A következő állítással tisztázta:
Úgy gondoljuk, hogy a 30X körüli (és 150X feletti) oldalakat az alacsony leképezési minőség miatt szűrjük (mivel ez az egész genom, így sok olyan terület van, amely nehéz feltérképezéséhez), amely megmagyarázza ezeken a területeken az alacsony érzékenységet. Amint a hibasávok mutatják, nagyon kevés változat van ezeken az alacsony feltérképezési minőségi területeken.
Ezután közzétette a következő ábrát, amely sokkal jobban beleillik abba, amire mindannyian számíthatnánk. Ez egy olyan diagram, amely érzékenységet mutat a NA12878 iránt a NIST igazsághoz képest. 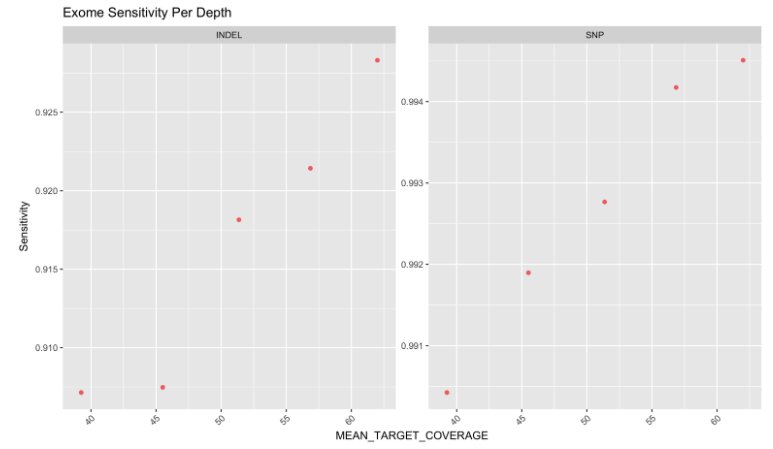
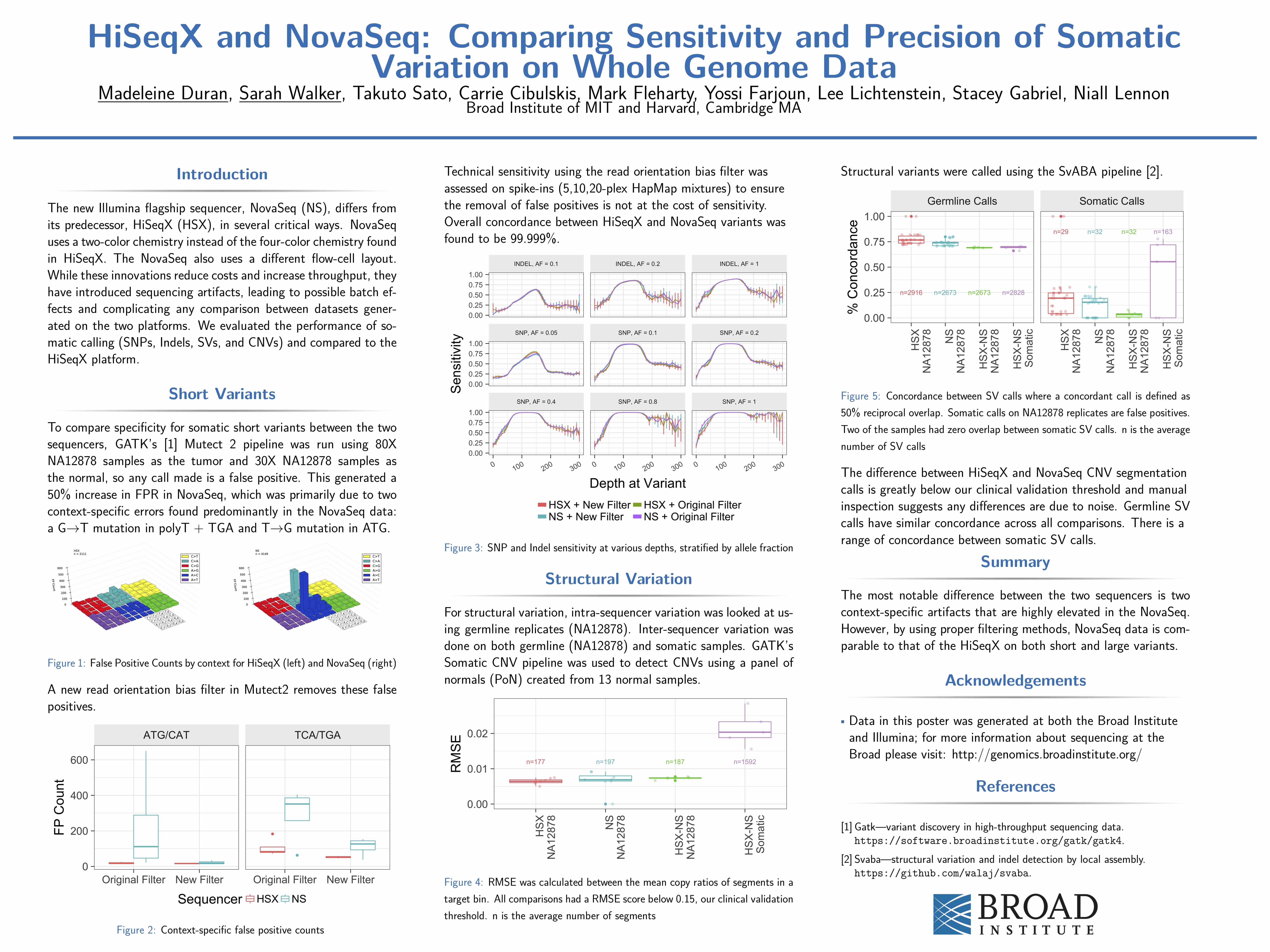
shlee, a GATK fórumaiból, rámutatott egy egy AGBT 2018 plakátra, ahol a szerzők összehasonlították a HiSeqX és a NovaSeq eszközöket. A 3. ábra az SNP-k és a rövid INDEL-ek iránti érzékenységet mutatja a tüskés bejutásokból származó több allélfrakción (AF). Meglepődve tapasztaltam, hogy az SNP-k, ahol az AF = 0,4 csak ~ 50% -os érzékenységet jelent ~ 25x lefedettségnél, és csak ~ 75% -os érzékenységet jelentenek ~ 50x lefedettségnél .
A poszter a szomatikus variációkra irányul, de a 3. ábra adatainak úgy kell viselkedniük, mint egy szokásos diploid csíra variáns, mivel gyakran látni az allél frakciók széles skáláját a csíra változatoktól. Spike-ineket használtak a 3. ábra számára. Nem világos számomra, hogy a 3. ábrán HaplotypeCaller-t használtak-e, de nem tudom, hogyan használhatták volna a Mutect2-t, mint az 1. ábrán & 2.
Ezen információk alapján úgy tűnik, hogy a standard 30x-os genom-lefedettség általában nem elegendő sok heterozigóta mutáció magabiztos hívásához, és ez a ~ 90-100x ideális.
Hiányzik valami? Tényleg hiányzik az a sok heterozigóta mutáció a standard WGS / WES-ben?
Egyelőre megválaszolatlanul hagyom ezt a kérdést, hogy mások is beleszólhassanak tolmácsolásba stb. Nagyra értékelném a további gondolatokat.